





Introduction
Degenerative neuropathy is a disorder characterized by reduced cognitive, walking, and athletic ability due to abnormal functioning of the brain or spinal cord caused by abnormal nerve cell death, unlike the normal aging process. The main cause of degenerative diseases is reported to be oxidative stress including that induced by reactive oxygen species (ROS) (Yoon et al., 2007). When ROS are generated in the body in an appropriate amount, they protect against pathogens or foreign substances by sterilization, whereas in excess, they slow down the physiological processes or kill the nerve cells (Cho and Sheen, 2009). Moreover, ROS induce damage proteins and lipid peroxidation, DNA, and cell membranes, resulting in various degenerative neurological diseases such as Parkinson’s disease, Alzheimer’s disease, and stroke (Ha and Park, 2006). Therefore, antioxidative activities play a crucial role in preventing damage to neuronal cells.
Moringa oleifera, a plant belonging to the family Moringaceae, is a widely grown species in the tropical and subtropical regions of Africa and Asia. The leaves of M. oleifera are particularly rich in various phytochemicals such as flavonoids, phytoestrogens, carotenoids, amino acids, minerals, vitamins, alkaloids, glycosides, sterols, caffeoylquinic acids, moringine, moringinine, and phenolics (Anwar et al., 2007; Fuglie, 1999; Guevara et al., 1999). In recent years, M. oleifera leaves, gums, flowers, seeds, and roots have been widely used to treat numerous diseases including inflammation, liver and cardiovascular diseases, regulating blood sugar, immune boosting agent, and cholesterol (Faizi et al., 1994; Limaye et al., 1995; Rao and Misra, 1998).
Several classes of flavonoids have been recognized to show antioxidant capacity against various oxidizable compounds. Flavonoids are rich in the plant kingdom and are common source in the flowering tissues, leaves, and pollen (Larson, 1988). Flavonoids in the plant are a crucial portion of the human diet due to their nutritional effects. These antioxidants suppress lipid peroxidation associated with carcinogenesis, thrombosis, and atherogenesis, and their known characteristics include inhibition of oxidative and hydrolytic enzymes (lipoxygenase, cyclooxygenase, and phospholipase A2), free radical scavenging, potent antioxidant capacity, and anti-inflammatory action (Pietta, 2000).
Numerous researchers have studied the bioactivity of M. oleifera by evaluating its antioxidant (Kwon and Youn, 2014), skin improving (Lee and Chang, 2012), and breast cancer inhibitory (Cho and Chang, 2014) activities. However, there is insufficient information on the neuroprotective and antioxidant effects of M. oleifera leaves ethanol extracts. Therefore, this study was carried out to evaluate the neuroprotective effects of M. oleifera leaf ethanol (80%) extract (MOLE).
Materials and Methods
Roswell Park Memorial Institute (RPMI) 1640 medium and fetal bovine serum (FBS) were obtained from Gibco BRL (Grand Island, NY, USA). Analytical grade of thiobarbituric acid (TBA), trichloroacetic acid (TCA), 2,4,6-tripyridyl-S-triazine (TPTZ), potassium persulfate, 2,2′-azino-bis(3ethylbenzthiazoline-6-sulfonic acid) (ABTS), Folin-Ciocalteu’s phenol reagent, hydrogen peroxide (H2O2), ferrous sulfate (FeSO4), kaempferol, quercetin, myricetin, streptomycin, penicillin, sodium bicarbonate, 2-[4-(2-hydroxyethyl) piperazin-1-yl] ethanesulfonic acid (HEPES), lactate dehydrogenase (LDH) assay kits, catechin, 3-[4,5-dimethythiazol2-yl]-2,5-diphenyl tetrazolium bromide (MTT), α-tocopherol, ascorbic acid (vitamin C), dimethyl sulfoxide (DMSO), and 2′,7′-dichlorofluorescein diacetate (DCFDA), were obtained from Sigma Chemical (St. Louis, MO, USA). All other reagents used were of the analytical grade available commercially.
Powdered M. oleifera Lam. leaf samples native to India were obtained from JK nutra (Gyeonggi, Korea) and were kept at –20°C until used. Freeze-dried leaf samples were prepared as follows: powdered M. oleifera leaves (50 g) were extracted with deionized water (di-water) and 80% ethanol (500 mL, 70°C) for 3 h. Whatman No. 2 filter paper (Whatman International Ltd., Kent, UK) was used to filter the extracts and evaporated under 40°C using a vacuum evaporator. The remaining extract was frozen, freeze dried, placed in a glass beaker, and kept at –70°C until used. The freeze dried extracts were re-dissolved in 10% DMSO to a concentration of 1.0 mg/mL before use.
Phenolic compounds were measured at 280 nm after separation using an Agilent high-performance liquid chromatography (HPLC) system (1100 series, Agilent, Santa Clara, CA, USA) (Wang and Helliwell, 2001). For flavonol determination, 1 g M.oleifera leaf material was combined with 20 mL 60% ethyl alcohol and 2.5 mL 6 M hydrochloric acid (HCl). Next, the hydrolyzed solution was refluxed at 95°C for 2 hours, filtered into a 50-mL volumetric flask, and then made up with 60% ethyl alcohol. Before injection into the Agilent HPLC system analysis, approximately 1 mL of the final solution was cooled under tap water and filtered through a 0.45-m filter (Nylon Acrodisc 13, Gelman, Ann Arbor, MI, USA). The analysis of flavonols (kaempferol, myricetin, and quercetin in the sample was performed using an Agilent 1100 series LC system, comprising a vacuum degasser, quaternary pump, auto sampler, thermostatted column compartment, and diode array detector. The column was a LiChrospher RP-18 column (250×4.6 mm i.d., 5-μm, Merck, Darmstadt, Germany). The mobile phase consisted of 30% acetonitrile in 0.025 M monopotassium phosphate (KH2PO4) buffer solution (v/v), and the pH was adjusted to 2.5 with 6 M HCl. The sample injection volume, column temperature, and flow rate were 20 μL, 30°C, and 1.0 mL/min, respectively. The ultraviolet (UV) spectra were measured at 200 and 400 nm, and 370 nm was measured as the peak area.
The PC12 cell line used in this study (KCLB 21721, Korea Cell Line Bank, Seoul, Korea) was obtained from a transplantable rat pheochromocytoma. Neuronal phenotype is induced reversibly by nerve growth factor. RPMI 1640 medium was used to cultivate PC12 cells that contained 100 μg/mL streptomycin, 50 units/mL penicillin, 25 mM sodium bicarbonate, 25 mM HEPES, and 10% FBS.
The intracellular ROS levels were determined using the DCFDA (fluorescent probe) assay (Heo et al., 2001). In brief, PC12 cells were plated in 96-well plates (104 cells per well) and treated for 15 min with the indicated concentrations of MOLE or ascorbic acid (vitamin C). After both treatment options were applied, the cells were incubated for 2 h in the presence of 200 μM H2O2 in phosphate buffered saline (PBS) or 50 μM DCFDA in PBS. A TECAN SERNR 94572 fluorometer (San Jose, CA, USA) was used to quantify the fluorescence at excitation and emission wavelengths of 485 and 530 nm.
The MTT assay was performed using an in vitro toxicology assay kit (TOX-1, Sigma-Aldrich). Ten μL of neuronal PC12 cells were grown (106 cells per well) in 96-well plates in 100 μL RPMI. Then the cells were pre-incubated with MOLE for 48 h before treatment with or without 200 μM H2O2 for 2 h. The amount of the MTT formazan product generated was determined by observing the absorbance of the reaction solution using a microplate reader (680, Bio-Rad, Tokyo, Japan) at test and reference wavelengths of 570 and 690 nm, respectively. Neuronal PC12 cells were centrifugated at 10,000×g for 5 min at 25°C, and then 100 μL of the supernatant was moved to new wells, followed by LDH determination using an in vitro toxicology assay kit (TOX-7, Sigma-Aldrich). By measuring the intracellular LDH released into the medium, we measured the damage to the plasma membrane.
The total phenolics were measured spectrophotometrically (Jeong et al., 2010). In brief, a 2-mL aliquot of the diluted extracts was placed in a 50-mL volumetric flask with 18 mL di-water. The reagent blank was prepared with deionized distilled water. Then, 2 mL Folin-Ciocalteu’s phenol reagent was transferred to the mixture, which was shaken and after 10 min, 20 mL of a sodium bicarbonate (Na2CO3) reagent was mixed. The combined mixture was directly diluted to 25 mL with deionized di-water, vortexed, left for 60 min at 25°C, and then the optical density was recorded at 750 nm. The standard curve of the total phenolic compounds was constructed using gallic acid standard solutions (0–250 mg/L) using the same protocol described above. The total phenolic compounds in the M. oleifera leaf water extract (MOLW) and MOLE were expressed as milligrams of gallic acid equivalents per 100 g of dried sample.
Seven mmol/L of ABTS·+ was prepared by dissolving in water. In order to create ABTS+, ABTS stock solution was reacted with 2.45 mmol/L potassium persulfate (final concentration) and allowed to stand at room temperature for 12-16 hours before use (Pellegrini et al., 1999). To test the samples, the ABTS·+ stock solution was mixed with 5 mmol/L of PBS. Final pH and optical density at 734 nm of ABTS·+ stock solution were 7.4 and 0.70, respectively. After adding 2.0 mL diluted ABTS·+ to 20 μL of the sample, the optical density was read 10 min after the combination. This activity is expressed as the percentage ABTS·+ scavenging and is calculated as follows:
where, Abs is absorbance.
This assay was performed according to a previous method (Chang et al., 2001). Young adult male Balb/c mice brains were dissected and homogenized in the Tris-HCl buffer (20 mM, pH 7.4), then the mixture was centrifuged at 5,000 rpm for 20 min at 5°C. Then, 1-mL supernatant was incubated with the samples in the presence of 0.1 mM vitamin C and 10 μM FeSO4 at 36°C for 2 h. The reaction was finished by the successive addition of 2.0 mL TCA (30%, w/v) and 3 mL TBA (1.5%, w/v), and then the mixture was boiled at 95°C. After 20 min, the color of the TBA-MDA mixture was observed at 532 nm and (+)-catechin was used as a reference. The MDA inhibition percentage was converted as follows:
Results and Discussion
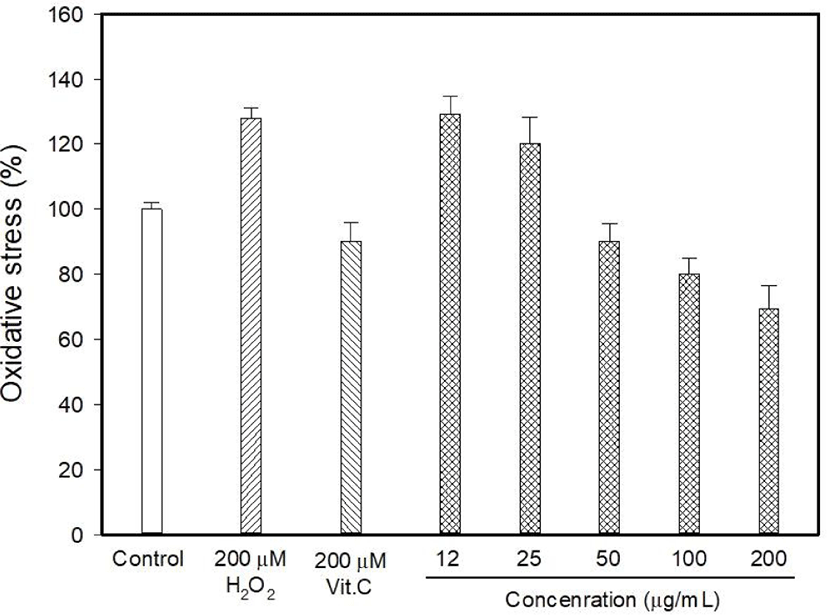
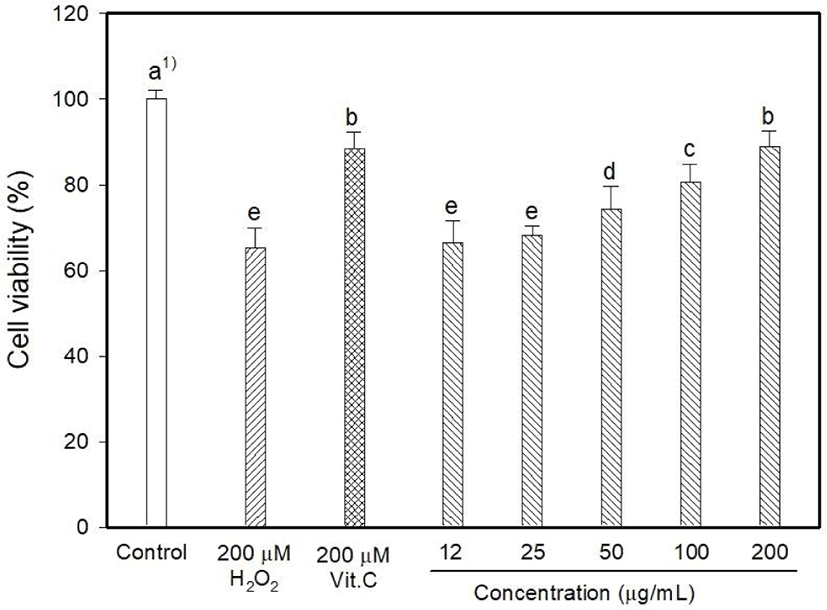
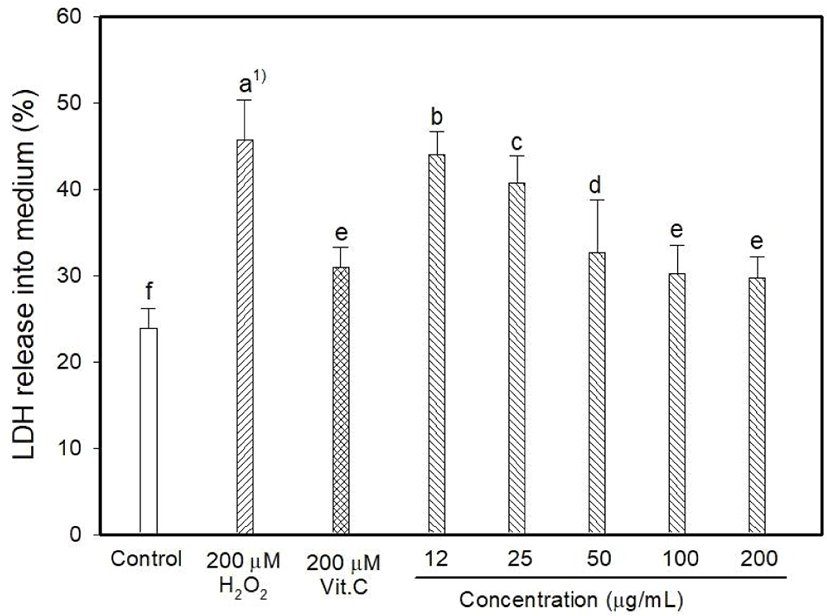
Myricetin and kaempferol were detected in the MOLE at concentrations of 5.44 and 1,157.13 mg 100/g, respectively (data are not shown). The determination of intracellular ROS generation using DCFDA formation showed that H2O2 induced 128.06% oxidative stress, whereas vitamin C reduced it to 90.26% (Fig. 1). DCFDA formation occurred at low levels at MOLE concentrations of 100 and 200 μg/mL than that observed with vitamin C (p<0.05) although it allowed the generation of a high level of DCFDA at concentrations of 12 to 25 μg/mL compared to that observed with vitamin C. In this study, the effect of MOLE on the survival of PC12 cells was examined using the MTT reduction assay, and it significantly prohibited the proliferation of PC12 cells in a dose-dependent manner (Fig. 2). In addition, similar levels of LDH were released into the culture medium (%) by vitamin C and MOLE at concentrations of 100 and 200 μg/mL (Fig. 3, p>0.05).
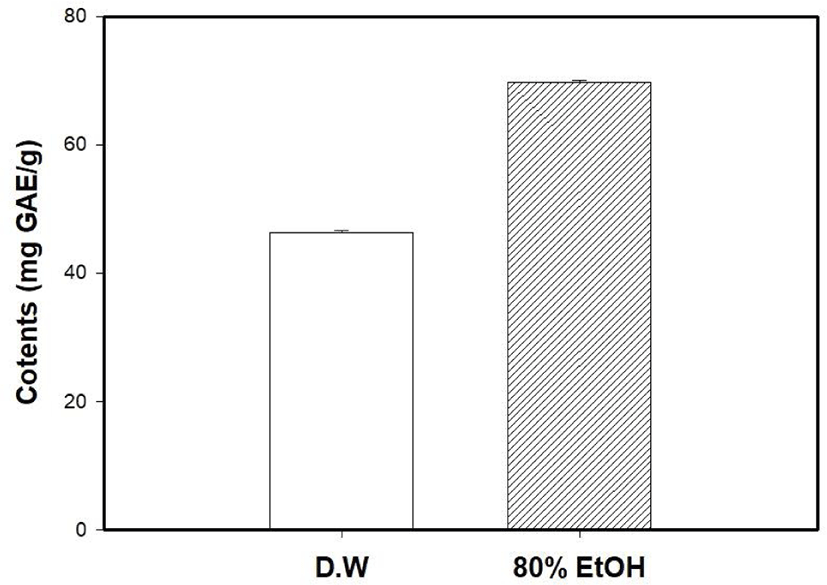
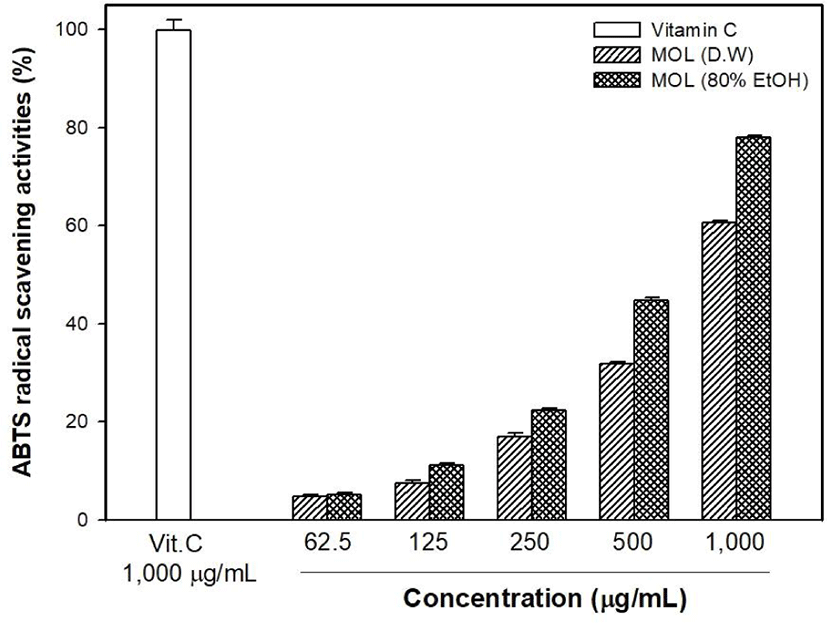
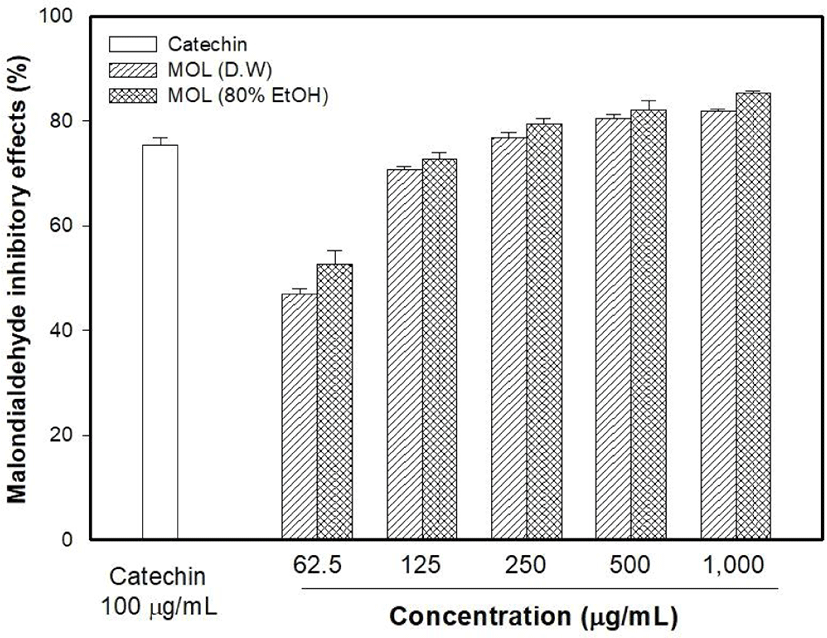
The antioxidant capacity of MOLE was measured and compared with that of the M. oleifera leaf water extract (MOLW). The total phenolic contents of the MOLW and MOLE were 46.33 and 69.75 μg/g, respectively (Fig. 4) and the ethanol extract showed a higher content than the water extract did (p<0.05). In the ABTS radical scavenging activity, the reference antioxidant agent, vitamin C (1,000 μg/mL), showed 99.86% activity (Fig. 5) while that of MOLW and MOLE ranged from 4.79 to 60.7 and 5.14 to 78.0 μg/mL, respectively and significantly increased in a concentration-dependent manner (p<0.05). No significant difference was observed at a concentration of 62.5 μg/mL between the treatments (p>0.05). However, the radical scavenging activity of MOLE was higher than that of MOLW at concentrations from 125 to 1,000 μg/mL. The MDA inhibitory effects of MOLW and MOLE ranged from 46.93% to 81.83% and 52.73% to 85.4%, respectively and MOLE showed higher MDA inhibition than MOLW did (Fig. 6, p<0.05).
The present study was carried out to evaluate the neuroprotective activity of MOLE, and the results clearly demonstrated that it showed significant effects by decreasing MDA levels, DCFDA formation, and LDH release into the cell culture medium while increasing cell viability. In addition, MOLE showed a higher antioxidant capacity than that of MOLW. In an Alzheimer’s disease rat, treated with M. oleifera leaves for 14 days enhanced learning and memory. Significant increases in antioxidant enzymes like catalase and superoxide dismutase were observed, while lipid peroxidase levels were greatly reduced (Ganguly et al., 2005). M. oleifera leaf extract, which is non-toxic even at high levels, was recently found to enhance memory by nootropic activity and the provision of substantial amounts of antioxidants such as vitamins E and C to combat oxidative stress (Ganguly and Guha, 2008; Mohan et al., 2005).
In addition, the protective effects of flavonoids on the neurons have been investigated, and the results show that flavonoids with high free radical scavenging efficacy defense neuronal cells against oxidative stress (Gao et al., 2001; Ni et al., 1996). The antioxidant characteristics of flavonoids guard hippocampal cells against the toxic mechanisms induced by A β peptides (Bastianetto and Quirion, 2002; Yang et al., 2001). Therefore, flavonoids would be expected to exert neuroprotective effects. Quercetin and kaempferol are principal phenols found in M. oleifera (Ganguly et al., 2005). In this study, both myricetin and kaempferol were detected in MOLE while kaempferol was the major flavonoid present. Molina-Jiménez et al. (2004) measured the protective effects of flavonoids on SH-SY5Y human neuroblastoma cells. They reported that 100 μM fraxetin showed a more potent protective effect against rotenone than that of myricetin (50 μM).
Conversely, Filomeni et al. (2012) reported that kaempferol protected primary neurons and SH-SY5Y cells from rotenone toxicity. Furthermore, they showed that kaempferol prevented propagation of apoptotic signals produced in response to oxidative stress, such as those produced by rotenone, by working on the p38MAPK and c-Jun-N-terminal kinase. A previous study showed that flavonoids could improve neurodegeneration and dysfunction in neurons (Youdim and Joseph, 2001) and we hypothesized that the neuroprotective activity of MOLE might be partly mediated by the flavonoid content of the extract, especially myricetin and kaempferol.
Thus, the antioxidant activity may be responsible for the observed neuroprotective activity in this study. In addition, the kaempferol concentration, which was more than 200 times higher than that of myricetin in the MOLE, likely induced the similar neuroprotective activity to that of vitamin C observed at concentrations of 100 μg/mL or higher. Therefore, the neuroprotective activity of MOLE is probably related to its antioxidant activity, and the effect of the solvent used should be considered in the extraction of polyphenol, which is necessary for the antioxidative capacity. The antioxidant activities of the plant material and extract yields are strongly influenced by the solvent used for extraction. Since plant materials contain antioxidant materials with varied chemical properties and polarities and, therefore, exhibit different solubilities in a particular solvent.
Polyphenols are commonly isolated from plant matrices with polar solvents. Solvents most suitable for this purpose are aqueous mixtures of methanol, ethanol, ethyl acetate, and acetone (Peschel et al., 2006). Siddhuraju and Becker (2003) evaluated the effects of three extraction solvents, water, 80% ethanol, and methanol on the concentrations of polyphenols and the antioxidant capacity of freeze-dried M. oleifera leaves. They reported that kaempferol and quercetin were the major flavonoids in lyophilized M. oleifera leaves, and quercetin was high in the methanol and ethanol extracts while kaempferol was higher in the water and ethanol extracts than it was in the methanol.
Conclusion
In this study, the antioxidant capacity of the MOLE was higher than that of MOLW. This result may be due to the high level of phenolic compounds in the MOLE than in the water extract. Therefore, to extract a higher amount of the total phenolic compounds from M. oleifera leaves and obtain more potent antioxidant effects, 80% ethanol would be a better extraction solvent choice than water would. In conclusion, MOLE showed higher antioxidative activity than MOLW did, which was likely because of a high flavonoid content, especially kaempferol. These antioxidative activities could be responsible for the neuroprotective effect of MOLE.