





Introduction
The increase in the incidence of obesity worldwide is due to greater relative decline in physical activity than increase in energy intake (Pescatello and Van Heest, 2000). Obesity can be attributed to environmental factors that promote physical inactivity and an excessive intake of calories and high-fat energy-dense foods, leading to a state of chronic positive energy balance (Hill and Melanson, 1999). Thus, environmental manipulation (e.g., changes in diet and physical activity) could alter or prevent some aspects of obesity (Rosenbaum et al., 1997). In an earlier study, Boozer et al. (1995) found a positive relationship between the percentage of kilocalories from fat in the diet and the percentage of body fat when rats were fed isocaloric diets. In most cases, obesity is probably due to subtle alterations in the interactions between genetic and environmental factors that favor the net deposition of calories as fat (Rosenbaum and Leibel, 1998). Moreover, high-fat overfeeding could lead to an accumulation of excess energy compared to repeated bouts of carbohydrate overfeeding (Hill et al., 2000). The hypothalamus is recognized as the main brain region regulating food intake as it is associated with caloric and nutrition requirements (Blouet and Schwartz, 2010; Volkow et al., 2011). Specifically, several limbic [nucleus accumbens (NAc), amygdala, and hippocampus] and cortical brain regions (orbitofrontal cortex, anterior cingulate cortexcingulate gyrus, and insula) and neurotransmitter systems (dopamine, serotonin, opioids, and cannabinoids), as well as the hypothalamus, are implicated in the rewarding effects of food (Petrovich et al., 2005). By contrast, the regulation of food intake by the hypothalamus appears to rely on the reward and motivational neurocircuitry to modify eating behaviors (Passamonti et al., 2009). Westerterp (2006) reported that energy intake from fat has a greater effect on body weight than energy intake from non-fat sources because excess energy consumption from the high-fat diet was greater in obese than in non-obese participants.
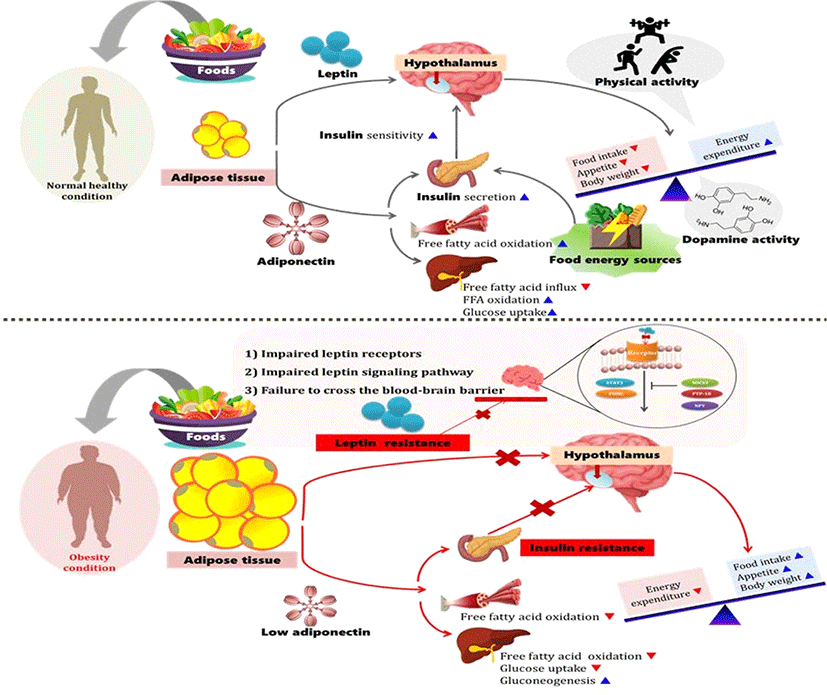
Thus, the author believes that energy sources (from lipid or carbohydrate) can influence the obesity epidemic since energy sources can largely affect physical activity after meals. Wang et al. (2009) reported that food ingestion is modulated by peripheral and central signals. Ghrelin increases food intake and body weight by stimulating neurons in the hypothalamus (Wang, et al., 2009). However, the relation between energy origins, physical activity, neurotransmitters, and hormones in obesity has not been fully studied. Therefore, the intent of this paper is to provide an overview of what is known and what is unknown about the effect of energy sources, physical activity, and endocrine substances on the prevention and treatment of obesity (Fig. 1).
Effect of food energy sources on obesity
The obesity epidemic is closely related to an imbalance between energy intake and energy expenditure. In other words, less energy being used compared to the energy intake from food results in weight gain. The central nervous system influences energy balance and body weight through three mechanisms: (1) effects of behavior, including feeding and physical activity; (2) effects on the activity of the autonomic nervous system, which regulates energy expenditure and other aspects of the metabolism, and (3) effects on the neuroendocrine system, including the secretion of hormones such as growth hormone, thyroid, cortisol, insulin, and sex steroids (Spiegelman and Flier, 2001). Diet is affected by metabolic, neural, and endocrine factors, and is modified by powerful visual, olfactory, emotional, and cognitive inputs (Spiegelman and Flier, 2001). Bodyweight management consists of body-weight control by promoting a stable balance between energy intake and energy expenditure (Westerterp-Plantenga, 2004). Westerterp-Plantenga (2004) reported that energy expenditure could be stimulated by increasing relative fat-free mass since this is the main determinant of energy expenditure. This means that fat intake was related to relatively lower satiety because the fat content of food appears to affect the meal frequency. Therefore, it is assumed that the accumulation of energy by fat intake increases, while the consumption of energy decreases. There is evidence that the consumption of a high-fat diet increases the total energy intake and that excess dietary fat is stored with greater efficiency than similar excesses of dietary carbohydrate or protein (Hill et al., 2000). Flatt et al. (1985) reported that dietary-induced thermogenesis is lower when a high-fat diet is consumed than when a low-fat diet is consumed. A greater proportion of excess energy is stored during fat overfeeding (~90-95%) compared with an equivalent amount of carbohydrate overfeeding (~75-85%) (Horton et al., 1995). This is because carbohydrate overfeeding has been reported to produce a progressive increase in carbohydrate oxidation and total energy expenditure, whereas fat overfeeding had minimal effects on fat oxidation and total energy expenditure (Hill et al., 2000). Moreover, the high caloric density and hydrophobic nature of triglycerides permit efficient energy storage without adverse osmotic consequences (Rosenbaum et al., 1997). Thus, these findings indicated that reducing dietary fat intake appears to be more effective in reducing weight than reducing dietary carbohydrate intake. Energy expenditure occurs from physical activity, basal metabolism, and adaptive thermogenesis (Spiegelman and Flier, 2001). This energy expenditure from physical activity could be influenced by dietary sources depending on the energy source. The feeling of satiety after eating may be due to the dietary source, which affects the level of physical activity after eating. However, various studies have reported the effects of food intake on obesity. Hall et al. (2015) demonstrated that calorie for calorie, restriction of dietary fat led to greater body fat loss than the restriction of dietary carbohydrates in adults with obesity. This occurred despite the fact that only the carbohydrate-restricted diet led to decreased insulin secretion and a substantial, sustained increase in net fat oxidation compared to the baseline energy-balanced diet (Hall et al., 2015). Hall et al. (2015) summarized that the selective reduction of dietary carbohydrates resulted in decreased insulin secretion, increased fat oxidation, and increased body fat loss compared to a eucaloric baseline diet. On the other hand, selective isocaloric reduction of dietary fat led to no significant changes in insulin secretion or fat oxidation compared to the eucaloric baseline diet; however, significantly more body fat was lost than that lost during a carbohydrate-restricted diet (Hall et al., 2015). Hu et al. (2018) reported that when C57BL/6 mice were exposed to 29 different diets varying from 8.3% to 80% fat, 10% to 80% carbohydrate, 5% to 30% protein, and 5% to 30% sucrose result in only increased dietary fat content was associated with elevated energy intake and adiposity. Low-carbohydrate diets have been increasingly used for weight reduction in recent years and have been heavily promoted in the media and scientific publications (Churuangsuk et al., 2018). In their systematic review, Churuangsuk et al. (2018) concluded that the low-carbohydrate diet vs. low-fat diet on weight loss show a substantial variation in methodological quality The reporting of adverse effects is scarce, and higher quality meta-analyses reported little or no difference in weight loss between the two diets. A review of various studies suggests that more in-depth research is needed to determine which food sources are closely related to obesity.
Effect of physical activity on obesity
Physical activity may be the most important factor for body composition regulation (Blanc et al., 2000). Energy expenditure due to physical activity may also be a regulatory signal for leptin production through several potential mechanisms, such as changes in insulin sensitivity, fatty acid concentrations, and alterations in sympathoadrenal activity (Blanc et al., 2000). Landt et al. (1997) reported that prolonged and strenuous exercise, such as marathon or ultramarathon training, may decrease leptin concentration. Obesity and physical inactivity are risk factors for the development of type 2 diabetes and other aspects of the metabolic syndrome and are attributed to insulin resistance (Kopelman, 2000). Physical activity, in conjunction with a weight loss program, could influence fasting patterns of lipid oxidation and contribute to improved insulin resistance (Goodpaster et al., 2003). Exercise also increases oxidative enzyme activity in skeletal muscles and induces related biochemical and morphologic changes that appear to confer a metabolic basis for improved insulin sensitivity (Goodpaster et al., 2003). Ross et al. (2000) found that weight loss induced by either caloric restriction or exercise improved insulin sensitivity by 60%, as compared with a 30% increase for exercise without weight loss. Goodpaster et al. (2003) suggested that the synergistic effect of weight loss and exercise could improve insulin resistance. Moreover, the combination of exercise and weight loss not only enhances the insulin-stimulated capacity for glucose utilization, but also enhances the capacity for fat oxidation during fasting conditions (Goodpaster et al., 2003). However, Malhotra et al. (2015) suggested that physical activity does not promote weight loss, and according to The Lancet’s global burden of disease reports, poor diet now causes more diseases than physical inactivity, alcohol intake, and smoking combined. They also suggested that members of the public are subjected to the unhelpful message of maintaining a ‘healthy weight’ through calorie counting, and many still wrongly believe that obesity is caused entirely due to lack of exercise. Some have explained that the extreme restriction of carbohydrate intake can reduce weight, but this also does not reveal the exact mechanisms underlying this process. Based on these studies, it is believed that the causes of obesity vary greatly depending on the level of exercise, type of diet, race, gender, age, and health conditions. Therefore, it is not only necessary to determine what the main cause of obesity is and how to treat it, but also to adopt a variety of approaches that can reduce the number of causes of obesity.
Effect of insulin on obesity
Insulin enters the brain from the circulation and acts to reduce energy intake and was the first hormonal signal to be implicated in the control of body weight by the central nervous system (Schwartz et al., 2000). There is evidence that brain insulin plays a role in feeding behavior, sensory processing, and cognitive function (Brody et al., 2009). Laboratory animals with disrupted brain insulin receptors show enhanced feeding (Bruning et al., 2000). Schwartz and Porte (2005) suggested that the brain plays a key role in the control of both body fat content and glucose metabolism. As body fat content and plasma glucose levels begin to increase, the circulating concentrations of leptin, insulin, and free fatty acids increase as well (Schwartz and Porte, 2005). In an earlier study, Wood et al. (1979) reported that food intake in a primate model decrease when a low dose of insulin is delivered directly to the brain by continuous intracerebroventricular infusion. When this fact was combined with evidence that insulin circulates at levels proportionate to body fat mass, that circulating insulin is transported into the brain, and that insulin receptors are concentrated in brain areas involved in the control of food intake and autonomic function, insulin emerged as a candidate “adiposity negative feedback” signal in the central control of energy homeostasis (Schwartz and Porte, 2005). Reduced neuronal insulin action increases food intake and body weight, and these effects may also cause insulin resistance (Schwartz and Porte, 2005). Cortisol may also contribute to enhanced lipolysis by further inhibiting the antilipolytic effect of insulin (Kopelman, 2000). These factors contribute to an exaggerated release of free fatty acids from abdominal adipocytes into the portal system (Kopelman, 2000). Insulin, as a neurotransmitter, stimulates neuronal glucose metabolism, regulates the glucose level in the blood vessels, and maintains body weight by maintaining a balance between insulin and leptin. Insulin, which is a regulator of appetite that rapidly increases after a meal, also varies directly with changes in adiposity and acts by inhibiting appetite (Austin and Marks, 2009). Unlike other neurotransmitters or hormones, the mechanisms underlying the effect of insulin on obesity seem relatively clear.
Effect of leptin on obesity
Leptin is a 16-kDa glycosylated protein consisting of 146 amino acids produced predominantly by adipose tissue. However, low levels of leptin expression were also detected in the hypothalamus, pituitary, placenta, skeletal muscle, and gastric and mammary epithelia (Moschos et al., 2002). Leptin was shown to directly target the central nervous system and supress appetite (Flier and Maratos-flier, 1998). In general, leptin is regulated by complex loops involving intake behavior, catecholamines, cortisol, growth hormone, and insulin (Mantzoros and Moschos, 1998). The mechanisms underlying leptin resistance in obese humans may include defective transport of leptin into the brain, or reduced hypothalamic leptin signaling, which is in part, due to the upregulation of specific inhibitors of leptin signaling (Gale et al., 2004). Leptin may play a role in many diverse physiologic processes, but it is primarily involved in energy homeostasis and satiety (Gale et al., 2004). Leptin is produced in adipocytes and is transported to the brain capillaries (Jing et al., 2004). Leptin levels in circulation increase in proportion to fat mass, and circulating leptin conveys information to the hypothalamus regarding the amount of energy stored in adipose tissue, suppresses appetite, and affects energy expenditure (Friedman and Halaas, 1998). Thus, leptin is currently highlighted as a protein of interest in obesity. For instance, leptin-deficient children exhibit ravenous feeding behavior and develop extreme obesity (Gale et al., 2004).
Earlier studies (Campfield et al., 1995; Rosenbaum et al., 1997) reported that systemic or intracerebroventricular administration of leptin reduces food intake and increases energy expenditure, resulting in reduced body fat and restoration of insulin-sensitive glucose disposal in ob/ob mice. Friedman and Halass (1998) reported that leptin administration to obese leptin-deficient animals reversed their hyperphagia, hypothermia, decreased locomotor activity, and all neuroendocrine and immunological abnormalities. Administration of leptin to fasting mice abrogates many of the neuroendocrine consequences of starvation, suggesting that the normal biological role of leptin may be to aid the organism in monitoring transitions between adequately nourished and starved states (Friedman and Halaas, 1998).
In another study, the administration of exogenous leptin to children resulted in a remarkable decrease in their energy intake and a dramatic loss of fat mass while maintaining a lean body mass (Farooqi et al., 2002). Van Heek et al. (1997) and El-Haschimi et al. (2000) reported that leptin injected directly into the brain suppressed food intake and induced hypothalamic signaling better than leptin injected via the peripheral route in mice with diet-induced obesity. However, direct administration of leptin into the central nervous system of ob/ob mice in doses that do not result in detectable levels in the periphery completely reverses the body weight and metabolic phenotype (Friedman and Halaas, 1998). However, Coll et al. (2007) reported that in the majority of cases of obesity, despite both an intact leptin receptor and high circulating leptin levels, leptin fails to bring about weight loss. Coll et al. (2007) reported that the precise mechanisms whereby increased fat stores signal the adipose tissue mass to produce more leptin remain mysterious; progress in this field of research has been impeded by the production of very low levels of leptin by the otherwise very useful adipocyte cell lines, using which the majority of the knowledge of adipocyte cell biology has been established (Coll et al., 2007).
Increase in plasma leptin concentrations in humans with obesity in directly proportional to the body fat mass. The reintroduction of energy supplies rapidly raises leptin levels and suppresses the starvation program (Spiegelman and Flier, 2001), and reduced leptin levels entrain a complex neural response characteristic of starvation that includes hunger and food-seeking behavior, efficient metabolism, and an array of neuroendocrine responses that favor survival during periods of limited energy (Ahima et al., 1996; Spiegelman and Flier, 2001). As fat mass increases, further rises in leptin levels have a limited ability to suppress food intake and prevent obesity, as seen by the prevalence of obesity despite high levels of circulating leptin (Considine et al., 1996).
A common form of obesity in humans occurs when high levels of leptin fail to suppress feeding and mediate weight loss; this is defined as leptin resistance (Myers et al., 2008). Leptin resistance in the hypothalamus invokes the starvation pathway and promotes food intake (Wang et al., 2009). Several mechanisms may contribute to leptin resistance, and reduced leptin-receptor signal transduction is another potential cause of leptin resistance (Woods et al., 2000). By decreasing the ability of circulating leptin to enter the brain interstitial fluid, where it can bind to neuronal leptin receptors, impaired leptin transport across endothelial cells of the blood-brain barrier is a potential mechanism underlying leptin resistance (Schwartz et al., 2000). Leptin is capable of effectively reducing food intake and body weight and was initially considered for use in the treatment of obesity (Izquierdo et al., 2019). However, endogenous leptin is not very stable in vivo (Izquierdo et al., 2019), and treating obese patients with exogenous leptin was unsuccessful in most cases since most patients already presented with high levels of circulating leptin and did not show any response to the treatment, which defined the so-called state of “leptin resistance” (Roujeau et al., 2014). Consequently, the treatment of obese patients with leptin was given less attention, and the focus of obesity research shifted toward the prevention and reversal of the state of leptin resistance (Roujeau et al., 2014). Nevertheless, the use of synthetic molecules similar to leptin, but ones that are more stable, may be helpful in the treatement of obesity. Novakovic et al. (2010) used a synthetic peptide amide with leptin‐like activity to show that intraperitoneal delivery of this peptide significantly ameliorates a number of metabolic dysfunctions associated with obesity in the ob/ob mouse model. The authors assume that further research is required to overcome leptin resistance and increase the safety of leptin so that it may be developed into a drug to treat obesity. Additionally, there is also a question of whether the development of drugs to treat obesity using leptin is necessary, because many drugs related to obesity treatment are already sold in the existing market.
Effect of adiponectin on obesity
Adiponectin is a 244-amino acid protein expressed and secreted exclusively from white adipose tissue; it acts as an insulin-sensitizing hormone whose blood concentrations are reduced in obesity and type 2 diabetes (Gale et al., 2004). Adiponectin is an adipocyte-derived hormone that has been proposed to play an important role in energy homeostasis (Trujillo and Scherer, 2005). Adiponectin was shown to be negatively correlated with body weight, body fat mass, and insulin levels in humans (Coll et al., 2007). In sharp contrast to leptin, plasma adiponectin levels are negatively correlated with body fat, decreasing with obesity and increasing in response to weight loss (Coll et al., 2007). Administration of recombinant adiponectin to rodents increases glucose uptake and fat oxidation in muscles, reduces hepatic fatty acid uptake and hepatic glucose production, and improves whole-body insulin resistance (Heilbronn et al., 2003). Nawrocki et al. (2006), using knockout mouse models, suggested that adiponectin is protective against the development of insulin resistance, glucose intolerance, and dyslipidemia. However, Coll et al. (2007) reported that the evidence suggesting that adiponectin has a role in controlling food intake is not convincing. Although the ectopic overproduction of adiponectin can reduce food intake in high-fat diet-fed rats and offset the development of diet-induced obesity (Coll et al., 2007; Shklyaev et al., 2003), food intake in adiponectin knockout mice is no different from that seen in wild-type littermates (Maeda et al., 2002). Adiponectin levels are reduced in obese individuals, particularly among patients with excess visceral adiposity (Cote et al., 2005). This is because adiponectin decreases food intake and obesity in obese rats and improves insulin sensitivity by decreasing hepatic glucose output (Austin and Marks, 2009). Achari and Jain (2017) suggested that the pharmacological elevation of circulating adiponectin will become a promising therapeutic strategy to ameliorate obesity-related diseases. Studies have found that supplementation of l-cysteine increases adiponectin secretion in adipocytes (Achari and Jain, 2017). Gomez-Arbelaez et al. (2013) reported that ingesting garlic extract for 12 weeks improved the adiponectin levels in patients with metabolic syndrome. In a mouse model of diabetes, treatment with cobalt (a heme oxygenase inducer) reduced visceral and subcutaneous obesity and increased insulin sensitivity through the upregulation of adiponectin (Li et al., 2008). Therefore, Achari and Jian (2017) suggested that drugs targeting adiponectin synthesis would be helpful in treating obesity, diabetes, and cardiovascular disease.
Effect of dopamine on food intake
Striatal dopamine signaling is regulated by several factors, including dopamine production, and has been implicated in obesity (Cone et al., 2013; Stice et al., 2008; Vucetic et al., 2012). Insulin released in response to ingested food has been shown to influence the dopamine transporter function. The dopamine transporter is one of the likely candidate targets for studying the effects of diet (Cone et al., 2013). Binding of dopamine to dopamine transporters, and hence, its availability, is reduced in high-fat diet-fed mice (Cone et al., 2013); high-fat diet-induced obesity in rat models is associated with a reduced rate of dopamine reuptake by dopamine transporters (Speed et al., 2011). In humans, ingestion of palatable food has been shown to release dopamine in the dorsal striatum in proportion to the self-reported level of pleasure derived from eating the food (Small et al., 2003). Upon first exposure to a food reward or an unexpected reward, the firing of dopamine neurons in the ventral tegmental area increases with a resulting increase in dopamine release in the NAc (Norgren et al., 2006; Volkow et al., 2011). Dopamine regulates food intake via the mesolimbic circuitry by modulating the appetitive motivational processes (Wise, 2006). Wang et al. (2009) reported that the dopamine increases were significantly correlated with the increases in self-reports of hunger and the desire for food. Palmiter (2008) suggested that the involvement of dopamine in the dorsal striatum seems to be crucial for enabling the motivation required to consume the food that is necessary for survival.
The mesencephalic dopamine system regulates pleasurable and motivating responses to food intake and stimuli (Volkow et al., 2008), which affects and alters behavioral components of energy homeostasis. Batterham et al. (2007) reported that the mesencephalic dopamine system could respond to food stimuli even in the presence of postprandial satiety factors. In addition, other mechanisms modulate eating behavior, such as stress, which increases the consumption of high energy density food, also contributing to obesity (Wang et al., 2009). The sensory processing of food and food-related cues plays an important role in the motivation for food, and it is especially important in the selection of a varied diet (Wang et al., 2009). Dopamine plays an important role in the tasting of palatable foods, which is mediated through the insula (Wang et al., 2009). Animal studies have shown that tasting sucrose increases dopamine release in the NAc (Hajnal et al., 2004; Wang et al., 2009). Subjects with obesity show a greater activation of the insula than normal controls when tasting a liquid meal that consisted of sugar and fat (DelParigi et al., 2005; Wang et al., 2009).
The amygdala is another brain region involved in eating behavior (Wang et al., 2009), and extracellular dopamine levels in the amygdala were increased in a preclinical study of food intake after a brief period of fasting (Petrovich and Gallagher, 2003). Smeets et al. (2008) also showed the activation of the amygdala with food-related stimuli, tastes, and odors. The amygdala is also involved with the emotional component of food intake, and stress-induced amygdala activation can be dampened by the ingestion of energy-dense foods (Dallman et al., 2003). Highly palatable foods can override internal homeostatic mechanisms through their action on brain dopamine pathways and may lead to overeating and obesity (Batterham et al., 2007). These studies indicate that not only does food intake affect the release of dopamine, but the release of dopamine also results in increased food intake. It is also thought that these findings are related to obesity because stress or psychological conditions may affect the secretion of dopamine and food intake.
Influence of meat consumption on obesity
Current knowledge on the influence of meat consumption on obesity is inconsistent because the relationship between them is not clearly established or may not be casual. Meat frequently gains a negative health image because it has high fat content and, in the case of red meat, is likely to promote various diseases such as diabetes, colorectal cancer, and obesity (Wang and Beydoun, 2009). Previous studies evaluated the association of meat intake with obesity risk and the results indicated that they have a positive relation (Larsson and Wolk, 2006). However, meat plays an important role as a rich source of protein and micronutrients, such as iron, selenium, vitamin A, and folic acid (Biesalski, 2005). The high protein and low carbohydrate contents in meat contribute to a low glycemic index, which is presumed to overcome the issues of overweight and insulin resistance that are related to obesity (Biesalski, 2005). Recent studies regarding weight loss reported that protein-rich or high-meat diets, low-carbohydrate diet, and restrictive high-fat diet resulted in better weight loss than diets with low-fat, low-protein, and low-carbohydrates. In addition, the high-meat and low-carbohydrate diets are responsible for prevention of obesity and diabetes. This result could be attributed to change in the body metabolism from utilization of glucose to utilization of stored body fat (Atkins, 2004; Bravata et al., 2003; Schonfeldt and Gibson 2008). Moreover, the odds ratios of overweight/obesity was lower in the no-meat diet as compared to those in the daily-meat-intake diet based on the Korea National Growth Charts, after being adjusted for confounding variables (Shin, 2017).
Therefore, the implication of these findings regarding the relationship between meat consumption and obesity is relatively limited. More research is needed to figure out the mechanisms implicated in obesity, especially those related to hormonal processes or signaling pathways that are involved in absorption of meat into the human body.